cluster (phytree)
Validate clusters in phylogenetic tree
Syntax
LeafClusters = cluster(Tree, Threshold)
[LeafClusters, NodeClusters]
= cluster(Tree, Threshold)
[LeafClusters, NodeClusters, Branches]
= cluster(Tree, Threshold)
cluster(..., 'Criterion', CriterionValue,
...)
cluster(..., 'MaxClust', MaxClustValue,
...)
cluster(..., 'Distances', DistancesValue,
...)
Input Arguments
Tree | Phylogenetic tree object created, such as created with
the |
Threshold | Scalar specifying a threshold value. |
CriterionValue | Character vector or string specifying the criterion to determine the number of clusters as a function of the species pairwise distances. Choices are:
|
MaxClustValue | Positive integer specifying the maximum number of possible clusters for the tested partitions. Default is the number of leaves in the tree. Tip When using the Tip When using the |
DistancesValue | Matrix of pairwise distances, such as returned by the |
Output Arguments
LeafClusters | Column vector containing a cluster index for each species
(leaf) in |
NodeClusters | Column vector containing the cluster index for each leaf
node and branch node in |
Branches | Two-column matrix containing, for each step in the algorithm, the index of the branch being considered and the value of the criterion. Each row corresponds to a step in the algorithm. The first column contains branch indices, and the second column contains criterion values. Tip To obtain the whole curve of the criterion versus the number
of clusters in |
Description
LeafClusters = cluster(Tree, Threshold)
Starting with two clusters (k =
2), selects the partition that optimizes the criterion specified by the'Criterion'propertyIncrements k by
1and again selects the optimal partitionContinues incrementing k and selecting the optimal partition until a criterion value =
Thresholdor k = the maximum number of clusters (that is, number of leaves)From all possible k values, selects the k value whose partition optimizes the criterion
[ returns
a column vector containing the cluster index for each leaf node and
branch node in LeafClusters, NodeClusters]
= cluster(Tree, Threshold)Tree.
[ returns
a two-column matrix containing, for each step in the algorithm, the
index of the branch being considered and the value of the criterion.
Each row corresponds to a step in the algorithm. The first column
contains branch indices, and the second column contains criterion
values.LeafClusters, NodeClusters, Branches]
= cluster(Tree, Threshold)
cluster(..., ' calls PropertyName', PropertyValue,
...)cluster with optional properties
that use property name/property value pairs. You can specify one or
more properties in any order. Enclose each PropertyName in
single quotation marks. Each PropertyName is
case insensitive. These property name/property value pairs are as
follows:.
cluster(..., 'Criterion', specifies the criterion to determine the number of
clusters as a function of the species pairwise distances. CriterionValue,
...)
cluster(..., 'MaxClust', specifies the maximum number of possible clusters
for the tested partitions. Default is the number of leaves in the
tree.MaxClustValue,
...)
cluster(..., 'Distances', substitutes the patristic distances in DistancesValue,
...)Tree with
a user-provided pairwise distance matrix.
Examples
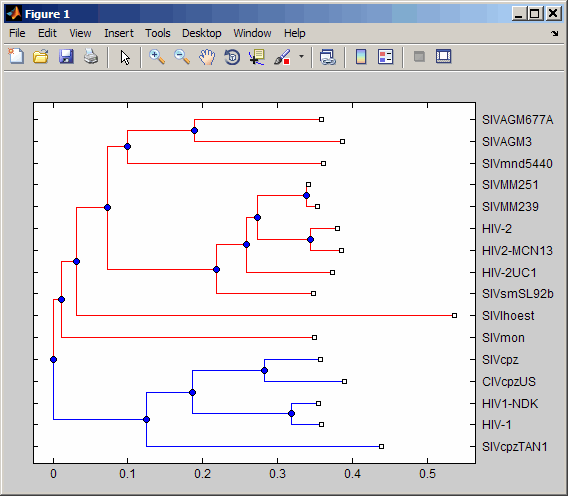
Validate the clusters in a phylogenetic tree:
% Read sequences from a multiple alignment file into a MATLAB
% structure
gagaa = multialignread('aagag.aln');
% Build a phylogenetic tree from the sequences
gag_tree = seqneighjoin(seqpdist(gagaa),'equivar',gagaa);
% Validate the clusters in the tree and find the best partition
% using the 'gain' criterion
[i,j] = cluster(gag_tree,[],'criterion','gain','maxclust',10);
% Use the returned vector of indices to color the branches of each
% cluster in a plot of the tree
h = plot(gag_tree);
set(h.BranchLines(j==2),'Color','b')
set(h.BranchLines(j==1),'Color','r')
References
[1] Dudoit, S. and Fridlyan, J. (2002). A prediction-based resampling method for estimating the number of clusters in a dataset. Genome Biology 3(7), research 0036.1–0036.21.
[2] Theodoridis, S. and Koutroumbas, K. (1999). Pattern Recognition (Academic Press), pp. 434–435.
[3] Kaufman, L. and Rousseeuw, P.J. (1990). Finding Groups in Data: An Introduction to Cluster Analysis (New York, Wiley).
[4] Calinski, R. and Harabasz, J. (1974). A dendrite method for cluster analysis. Commun Statistics 3, 1–27.
[5] Hartigan, J.A. (1985). Statistical theory in clustering. J Classification 2, 63–76.
See Also
cluster | phytree | phytreeread | phytreeviewer | plot | seqlinkage | seqneighjoin | seqpdist | silhouette | view