molviewer
Display and manipulate 3-D molecule structure
Syntax
molviewer
molviewer(File)
molviewer(pdbID)
molviewer(pdbStruct)
FigureHandle =
molviewer(...)
Input Arguments
File | Character vector or string specifying one of the following:
The referenced file is a molecule model file, such as a Protein Data Bank (PDB)-formatted file (ASCII text file). Valid file types include:
|
pdbID | Character vector or string specifying a unique identifier for a protein structure record in
the PDB database. Note Each structure in the PDB database is represented by a
four-character alphanumeric identifier. For example,
|
pdbStruct | A structure containing a field for each PDB record, such as
returned by the getpdb or pdbread function. |
Output Arguments
FigureHandle | Figure handle to the Molecule Viewer. |
Description
molviewer opens the Molecule
Viewer app. You can display 3-D molecular structures by selecting File
> Open, File > Load PDB ID,
or File > Open URL.
molviewer( reads
the data in a molecule model file, File)File,
and opens the Molecule Viewer app displaying the 3-D molecular structure
for viewing and manipulation.
molviewer( retrieves
the structural data of a protein, pdbID)pdbID,
from the PDB database and opens the Molecule Viewer app displaying
the 3-D molecular structure for viewing and manipulation.
molviewer( reads
the data from pdbStruct)pdbStruct, a structure containing
a field for each PDB record, and opens the Molecule Viewer app displaying
a 3-D molecular structure for viewing and manipulation.
FigureHandle =
molviewer(...)
Tip
You can pass the FigureHandle to
the evalrasmolscript function,
which sends RasMol script commands to the Molecule Viewer window.
Tip
If you receive any errors related to memory or Java® heap space, try increasing your Java heap space as described at https://www.mathworks.com/support/solutions/en/data/1-18I2C/.
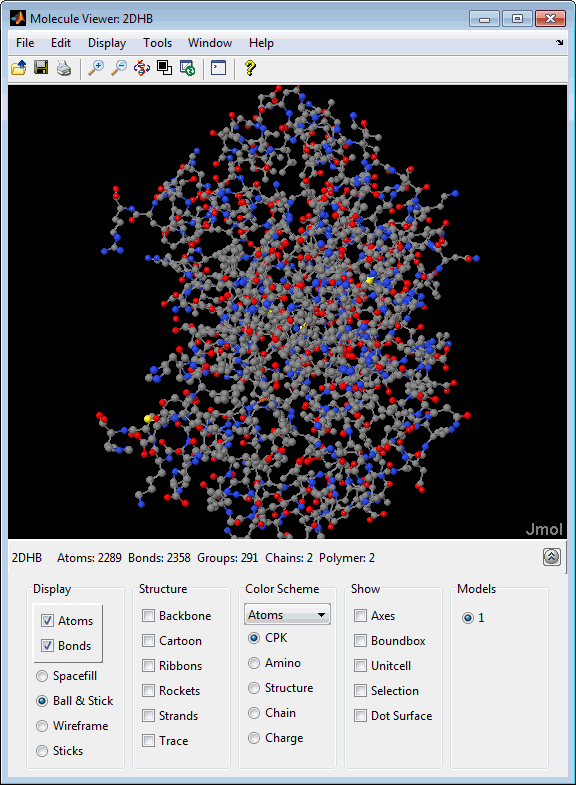
After displaying the 3-D molecule structure, you can:
Hover the mouse over a subcomponent of the molecule to display an identification label for it.
Spin and rotate the molecule at different angles by click-dragging it.
Spin the molecule in the x-z plane by clicking
 .
.Spin the molecule in the x-y plane by pressing and holding the Shift key, then click-dragging left and right.
Zoom in a stepless fashion by pressing and holding the Shift key, then click-dragging up and down.
Zoom in a stepwise fashion by clicking the figure, then turning the mouse scroll wheel, or by clicking the following buttons:
 or
or 
Move the molecule by pressing and holding Ctrl + Alt, then click-dragging.
Change the background color between black and white by clicking
 .
.Reset the molecule position by clicking
 .
.Show or hide the Control Panel by clicking
 .
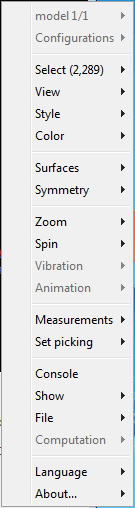
.Manipulate and annotate the 3-D structure by selecting options in the Control Panel or, for a complete list of options, by right-clicking the Molecule Viewer window to select commands:
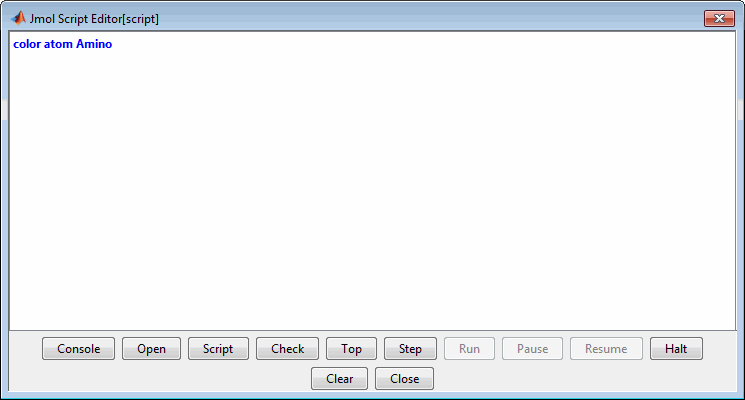
Display the Jmol Script Console by clicking
 .
.
Note
There is a known bug with the Open button of the script editor that prevents loading a Rasmol script interactively. Instead use the
evalrasmolscriptfunction which sends RasMol script commands to the Molecule Viewer app. Also, you can copy and paste the script commands into the script console.
Examples
View the acetylsalicylic acid (aspirin) molecule, whose structural
information is contained in the Elsevier MDL molecule file aspirin.mol.
molviewer('aspirin.mol')View the H5N1 influenza virus hemagglutinin molecule, whose structural information is located
at www.rcsb.org/pdb/files/2FK0.pdb.gz.
molviewer('http://www.rcsb.org/pdb/files/2FK0.pdb.gz') View the molecule with a PDB identifier of 2DHB.
molviewer('2DHB')View the molecule with a PDB identifier of 4hhb,
and create a figure handle for the Molecule Viewer.
FH = molviewer('4hhb')Use the getpdb function
to retrieve protein structure data from the PDB database and create
a MATLAB structure. Then view the protein molecule.
pdbstruct = getpdb('1vqx')
molviewer(pdbstruct) Compatibility Considerations
See Also
evalrasmolscript | getpdb | pdbread | pdbsuperpose | pdbtransform | pdbwrite